Данная патология означает нарушение питания мышц. Указанная патология является наследственной слабостью мышц, попадается крайне редко.
Этот синдром обычно мучает лишь мальчиков. 1 заболевание приходится на 3000 случаев рождения нормальных малышей.
Имеются другие разновидности мышечных дистрофий, которые возникают у девочек, но там проявления легче.
В ходе данного заболевания, при прогрессировании его возникает нарушение связей между мышцами и нервами.
Синдром Дюшена передаётся по наследству, мать - является носителем гена описываемой патологии, но не болеет сама.
Другие виды дистрофии
Кроме описываемого синдрома, имеются и другие типы данных дистрофий, хотя они редко возникают.
Синдром Беккера - крайне редок, и так же болеют лишь мужские особи. Патология возникает в 10-11 лет, и становится менее заметной в 35-40 лет.
Наследственная мускульная миопатия - заболевают и девочки и мальчики, так же имеет генетическую причину, наблюдается даже реже, чем описываемый синдром.
Плече-лопаточно-лицевая миопатия - крайне долгое развитие, течение благоприятное. Появляется до 10 лет. Характеризуется: будучи в грудном возрасте не могут хорошо высосать грудь; в более позднем возрасте не получается сделать губы трубочкой; тяжело осуществлять подъем рук, лицо имеет маскообразный вид.
Дистрофия Эмери—Дрейфуса - проявляется, как и остальные типы дистрофий, но в отличие от предыдущих, эти плохо влияют на сердце.
Первые симптомы
Ребёнок позже ходит, и то, все его попытки оказываются не успешными. Ребёнок ходит, переваливаясь из одной стороны в другую, часто падает на попу, желание подняться и встать из положения тела, сидя на полу, зачастую оказываются неудачными.
Мускулы ног могут выглядеть довольно крепкими, хотя в реальности таковыми не являются. Все остальные мышцы, отвечающие за ходьбу - развиты плохо.
Диагностика
Видя характерные симптомы, врач может подозревать у малыша данную патологию. В таком случае терапевт должен дать направление ребенку к ортопеду.
Исследование крови: в нормальной мышечной материи имеется креатин фосфокиназа. При данной патологии количество этого фермента слишком завышено.
Мышечный тест: при помощи электронного исследования мышц меряется быстрота передачи нервных реакций в мускулы.
Биопсия мышц: кусочек мышечной ткани рассматривается под микроскопом. При таком исследовании выявляются наросты в виде атеросклеротических бляшек.
Почему эта патология считается генетической
Указанный синдром, генетическое расстройство, появляющиеся из-за видоизменённой аллели в половой Х-хромосоме.
Любая клеточка человеческого организма, за исключением гамет содержит 46 хромосом. Одна аутосома содержит в своём составе множество алелий (около 1000). В аллелях и аутосомах расположена дезоксирибонуклеиновая кислота, ответственная за передачу данных, переходящих из одного поколения в другое.
Структура гена белковая. Глобулины — строительный материал для человеческого организма.
Данная болезнь появляется из-за нахождения специфического гена в Х-хромосоме, которая находится как в мужском, так и в женском организме. Аллель реагирует на продуцирование белка, формирующего нормальную мускульную ткань.
Девочки иногда получают в наследство изменённый аллель, но обычно, болезнь Дюшена у них не появляется, так как в их организме имеются две Х-хромосомы, одна из них, здоровая, замещает нарушение во второй.
В ходе определённых экспериментов были получены следующие данные, болезнь Дюшена у мальчиков бывает врожденной, и приобретенной из-за мутаций, случившихся в генотипе после рождения.
Прогрессирование патологии
С прогрессированием патологии, признаки выражаются более отчётливо, это происходит потому, что мышцы уже практически не могут способствовать адекватному перемещению. Со временем мышцы рук ослабевают, и малышу очень трудно становится брать и держать предметы. Мускулы рук и ног становятся дистрофичными, суставы приобретают тугоподвижность. Не редко возникает деформация локтевых, тазобедренных и коленных суставов. Мускулы, удерживающие позвоночный столб, перестают расти, из-за чего позвоночник приобретает изгибы. Малышу сложно ходить. Кой-какие дети плохо учатся. Научение вызывает кропотливый труд, в ходе чего ребёнок просто не успевает за учебной программой.
Как помочь малышу?
К сожалению, этот синдром не лечиться, поэтому стоит подсказать ребёнку, как адаптироваться к жизни с данной болезнью.
Ребёнку показана психотерапия, дабы убедить его в важности занятий физической нагрузкой.
Стоит упражняться в занятиях специальной гимнастикой, дабы сохранить движения в суставах.
Чтобы не образовывались контрактуры, стоит применять специальные шины и корсеты.
Показана лечебная ходьба.
Дети с указанным синдромом, испытывают некоторые трудности в научении, поэтому им необходим индивидуальный подход и внимание. Хорошо, если больной малыш станет учиться в специальной школе, для детей с данными трудностями. Существуют такие школы -интернаты, программа в которых адаптирована именно для такого контингента.
Если у больного малыша имеются братья или сёстры, им необходимо в равной степени уделять внимание, чтоб дети не думали, что они брошены.
Миодистрофия Дюшенна (МДД) - наследственное заболевание, которое начинается в возрасте 2-5 лет и характеризуется прогрессирующеймышечной слабостью, атрофией и псевдогипертрофией проксимальных мышц , нередко сопровождается кардиомиопатиями и нарушением интеллекта. На ранних этапах заболевания наблюдается повышенная утомляемость при ходьбе, изменение походки («утиная походка»). При этом происходит постепенная деградация мышечных тканей. 95% больных перестают ходить в возрасте 8-12 лет. В возрасте 18-20 лет больные, как правило, умирают, часто от дыхательной недостаточности. Выделяют аллельную МДД форму - мышечную дистрофию Беккера (МДБ, OMIM ), которая характеризуется сходными клиническими проявлениями, более поздним началом (примерно в 10-16 лет) и более мягким течением. Такие больные часто сохраняют способность ходить до 20 лет, а некоторые - до 50-60 лет, хотя в патологический процесс вовлечены те же мышцы, что и при МДД. Продолжительность жизни таких больных сокращена незначительно.
Биохимическим маркером заболевания является повышенный (в 100-200) раз уровень креатинфосфокиназы (КФК ) в крови. У носительниц поврежденного гена уровень КФК в среднем также несколько повышен.
Тип наследования мышечной дистрофии Дюшенна - Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена DMD, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Мышечная дистрофия Дюшена (МДД) встречается приблизительно у одного из 2500-4000 новорожденных мальчиков.
Ген DMD, ответственный за прогрессирующую мышечную дистрофию Дюшена/Беккера (МДД/МДБ), находится в локусе Хр21.2, имеет размер 2,6 млн п.н. и состоит из 79 экзонов. В 60% случаев мутации, приводящие к МДД/МДБ, представляют собой протяженные делеции (от одного до до нескольких десятков экзонов), в 30% случаев - точковые мутации и в 10% случаев - дупликации. Из-за наличия так называемых «горячих участков» делеций амплификация 27 экзонов и промоторной области гена DMD позволяет выявлять примерно 98% всех крупных делеций. Поиск точковых мутаций затруднен из-за большого размера гена и отсутствия мажорных мутаций.
В Центре Молекулярной Генетики проводится измерение уровня КФК в крови, а также прямая диагностика МДД/МДБ, представляющая собой поиск крупных делеций/lдупликаций во всех экзонах гена DMD и поиск «точковых» мутаций гена DMD методом NGS (next generation sequensing). Исследование методом NGS позволяет так же выявлять делеции всех экзонов гена DMD у больных мальчиков. Анализ всех экзонов гена позволяет определить точные экзонные границы делеции в сучае ее выявления, и таким образом, установить приводит ли данная делеция к сдвигу рамки считывания белка, что в свою очередь важно для прогноза формы заболевания - миодистрофия Дюшенна или Беккера. Таким образом, сочетание различных методов исследования позволяет выявлять практически все мутации гена DMD.
Наличие любого типа мутаций (делеции/дупликации в одном или нескольких экзонах, «точковые» мутации) является молекулярно-генетическим подтверждением клинического диагноза миодистрофии Дюшена/Беккера и позволяет проводить дородовую диагностику в данной семье.
Внимание! Для измерения уровня КФК кровь должна быть свежей (не замороженной)!
В случае дородовой диагностики необходим биоматериал плода, в качестве которого можно использовать ворсины хориона (с 8-й до 12-й недели беременности), амниотическую жидкость (с 16-й до 24-й недели беременности) или пуповинную кровь (с 22-й недели беременности).
Нами разработаны . Наборы предназначены для использования в диагностических лабораториях молекулярно-генетического профиля.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1 . Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Однако женщины являются носительницами дефектного гена, который провоцирует развитие симптомов. Первые проявления болезни могут быть замечены в возрасте от 2 до 5 лет, однако проявлений расстройства может не быть вплоть до 10 лет. Дети, которые больны миопатией, чаще падают и тяжелее поднимаются по сравнению со здоровыми сверстниками. При попытке встать ребенок сначала поднимается на четвереньки, затем , опираясь руками и . К 8-10 годам у пациента могут начаться проблемы с походкой. Болезнь начинает прогрессировать и примерно к 10-12 годам мальчик может утратить способность передвигаться .
В результате генетического расстройства формируется сколиоз, наблюдаются повреждения сердечного миокарда, что становится заметно на ЭКГ. В среднем больные миопатией Дюшена погибают в возрасте 20 лет, однако существуют случаи, когда пациенты доживают до 25-30 лет. Причиной смерти становится сердечная и легочная недостаточность в результате мышечной атрофии.
Лечение
Заболевание диагностируется путем анализа ДНК. Также можно обнаружить миопатию, исследовав состав мышечной ткани пациента на наличие дистрофина. Второй метод чаще используется для подтверждения диагноза, а первый дает возможность установить факт наличия конкретной формы болезни.
Препаратов для лечения миопатии Дюшена не существует. На сегодняшний день разрабатываются лекарства, позволяющие снизить скорость развития заболевания. Консервативное лечение направлено на контроль появляющихся в процессе симптомов с целью улучшить качество жизни. Как правило, стандартная терапия включает в себя назначение кортикостероидных лекарств («Преднизолон» или «Дефлазакорт»), которые позволяют увеличить энергию и силу пациента, а также уменьшить интенсивность симптомов.
Некоторые исследования показали, что мышечную силу у пациентов можно увеличить при помощи бета-2 агонистов («Сальметерол», «Формотерол»), однако данные препараты не замедляют течение болезни. Физическая активность (например, плавание) делают прогрессирование заболевание более плавным, а постельный режим, наоборот, ухудшает состояние больного.
Терапия через физические упражнения помогает сохранить мышечную функцию, а ортопедические устройства (например, шины, трости или инвалидные кресла) помогают улучшить качество жизни и дать возможность пациенту самому себя обслуживать. На последних стадиях болезни важна дыхательная поддержка пациента в условиях стационара при помощи специальных вспомогательных приборов.
Генные болезни - это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена.
Мышечная дистрофия Дюшенна (МДД) - это серьезное рецессивное заболевание, сцепленное с Х-хромосомой, которое характеризуется быстрым прогрессированием мышечной дистрофии, которая в конечном итоге приводит к полной потере способности двигаться и смерти больного.
Обычно на МДД болеют только мужчины, хотя женщины могут иногда быть носителями заболевания. Если же отец болен МДД, а мать является носителем, или тоже больна, то в таком случае на мышечную дистрофию Дюшенна может заболеть женщина. Расстройство возникает в связи с мутацией в гене дистрофин, который у людей расположен на Х-хромосоме (Xp21).
Симптомы заболевания обычно появляются у детей мужского пола до 5 лет и могут проявиться в раннем детстве. Первыми признаками болезни являются прогрессирующая проксимальная слабость мышц ног и таза, связанная с потерей мышечной массы. Постепенно эта слабость распространяется на руки, шею и другие части тела. В процессе прогрессирования заболевания мышечная ткань постепенно заменяется жировой и фиброзной тканями. Для помощи при ходьбе, в возрасте 10 лет может быть необходимым применение специальных подтяжек, но большинство пациентов уже старше 12 лет не могут передвигаться без инвалидной коляски. Позже возникают следующие признаки расстройства: аномалии развития костей, которые приводят к деформации скелета, в том числе к искривлению позвоночника.
Диагностика: ДНК-тест, биопсия мышц, пренатальное тестирование.
Лечение
Никаких известных эффективных препаратов для лечения мышечной дистрофии Дюшенна, не существует. Хотя согласно последним исследованиям стволовых клеток существуют перспективные векторы, которые могут заменить поврежденные мышечные ткани. Однако, на данном этапе лечения, как правило, симптоматическое и направлено на улучшение качества жизни больного человека.
43. Основные принципы генетического консультирования.
Первым условием генетического консультирования является уверенность в том, что поставленный диагноз правильный.
После установления диагноза в процессе генетического консультирования следует:
1 - решить, должны ли присутствовать при беседе оба родителя (подросткам необходимо предоставлять возможность для отдельной беседы);
2 - обсудить медицинские последствия дефекта; если это имеет отношение к делу, необходимо объяснить возможную вариабельность проявлений и течения заболевания и исходы, которые можно ожидать в будущем;
3 - проанализировать генетический анамнез каждого из родителей и выявить нераспознанный генетический риск;
4 - проанализировать интерпретации членов семьи или предоставленные другим лицом;
5 - описать консультируемым генетические основы заболевания, используя наглядные пособия (картины, демонстрирующие фенотип или другие признаки заболевания, изображения хромосом, диаграммы типов наследования);
6 - объяснить риск развития генетического заболевания в терминах, понятных семье;
7 - очертить круг возможного выбора решения: иметь детей, принимая во внимание возможный риск, не иметь детей, усыновить ребенка, если это возможно, провести искусственное обсеменение (это решение особенно уместно при всех случаях аутосомно-рецессивных заболеваний и серьезных аутосомно-доминантных заболеваний со стороны отца), отметить, возможно ли провести пренатальную диагностику;
8 - предложить консультируемым итог проведенного обсуждения, и, если возможно, встретиться с ними еще раз для того, чтобы помочь решить, как лучше всего поступить в данном случае;
9 - поддерживать контакт с семьями, проконсультированными ранее, для ознакомления их с информацией, которая может быть полезна, например с новыми методами выявления носительства у родителей или пренатальной диагностики.
В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.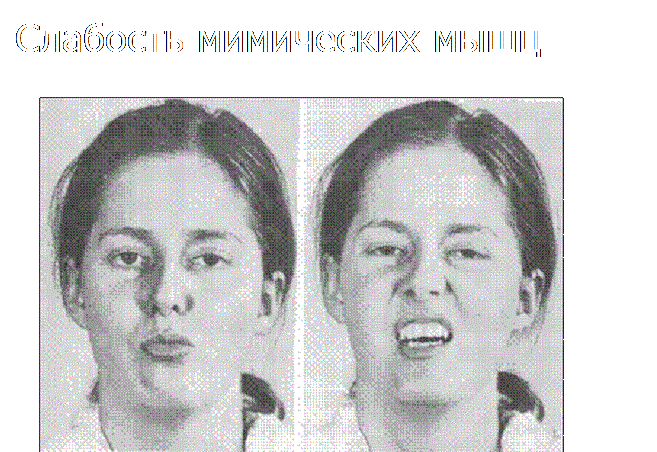
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!